Unit - 3
Miscellaneous Topics
Introduction
Molecular symmetry deals with the classification and the description of the molecule symmetry utilizing both mathematical and the general considerations of symmetry.
The group theory and symmetry give us the efficient tools to describe the geometry of objects from the details of the patterns in their structure. In chemistry the concept of chemistry helps to describe the chemical bond types precisely that occur between atoms or group of atoms in molecules, it also describes the transitions between energy levels in molecular systems, which contributes to the understanding of the absorption properties of the molecules and thus their spectra.
Molecular Symmetry is intended to introduce the concept of combining symmetry with spectroscopy clearly and has made it easily accessible. Designed to introduce the subject by combining symmetry with spectroscopy in a clear and accessible manner. It’s a modern and broad introduction to the various levels of symmetry.
Symmetry elements and Symmetry operations
A symmetry is a geometrical entity such as a line, a plane, or a point about which one can perform an operation of rotation reflection or inversion.
A symmetry operation is movement of a molecule or object about a symmetry element such that the resulting configuration is indistinguishable from the original.
A symmetry operation will transform a molecule into an equivalent or ideal configuration.

1. Identity E
2. Axis of symmetry [Cn]:
For e.g., water molecule
Principal and Subsidiary Axes:
3.Plane of Symmetry 
Classification of mirror plane:
Inversion centre of symmetry:
Improper axis of symmetry or rotation-reflection axis or alternate axis of symmetry:
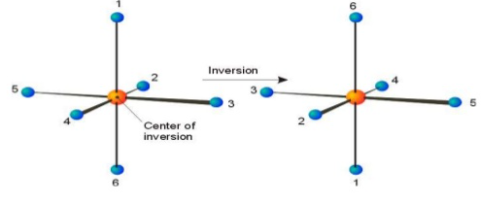
Centre of Symmetry
A Centre of symmetry is a point present in space of any group on the molecule that can be reflected back through the same point, but in the opposite direction at an equal distance, resulting in the formation of an equivalent group.
It is a point within the molecule from which lines drawn to opposite direction meet similar points at exactly the same distance and direction
It is also a line or point present within the molecule, if a straight line is drawn from any part of the molecule that point and extended an equal distance by a straight line on the other side, a like atom or part of the molecule is encountered. Centre of symmetry is also known as inversion Centre. This may be illustrated by the following example.
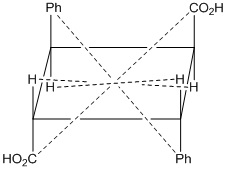
Fig: 1 α- Truxillic acid
If the Centre of inversion is considered as origin of a cartesian co-ordinate system, then the operation of inversion is to change the point of co-ordinates x, y, z. into a point of coordinates –x- y- z.
It is a point within the molecule from which lines drawn to opposite direction meet similar points at exactly the same distance and direction
A centre of symmetry is an imaginary point present in the molecule, so that if a line is drawn from any group of the molecule to this point and extended to an equal distance beyond the point, it meets the mirror images of the original group.
Centre of symmetry is also called centre of inversion, denoted by Ci. For example, 2,4-dimethyl cyclobutane-1,3-di-carboxylic acid exhibit centre of symmetry and so it is optically inactive.
Fig: 2,4-dimethyl cyclobutane-1,3-di-carboxylic acid
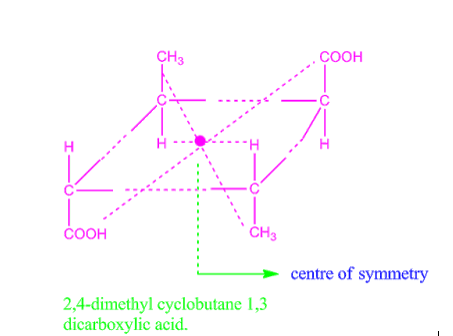
Fig: Centre of symmetry in 2,4-dimethyl cyclobutene 1,3 dicarboxylic acid
Why cis form of 2,5-dimethyldihydropyrimidine-4,5-dione is optically active, but trans form optically inactive?
The cis form of 2,5-dimethyldihydropyrimidine-4,5-dione has no centre or plane of symmetry, so it is optically active whereas trans form of 2,5-dimethyldihydropyrimidine-4,5-dione has a centre of symmetry, hence it is optically inactive.
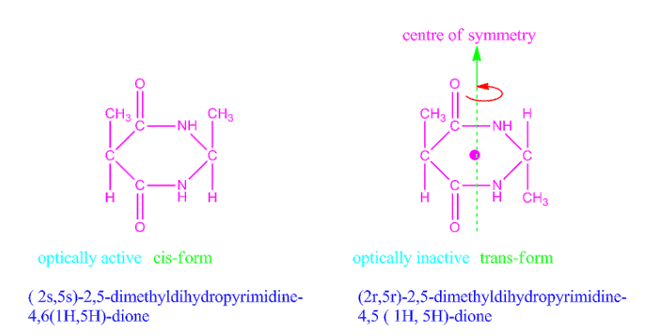
Fig: Cis and trans forms
Axis of Symmetry and Plane of Symmetry
Axis of symmetry is a line that splits the object into to equal halves, resulting in the creation of mirror image or mirror-like reflection on either side of the object. The word symmetry indicates balance, the concept of symmetry can be applied to various situations and contexts. Symmetry is a key concept in geometry which cuts the figure into two halves that are exact reflections of each other, as shown in the figure given below.
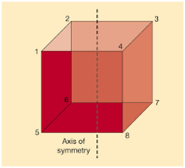
Fig: Axis of symmetry in a cube
For a parabola, the axis of symmetry is given by the formula,
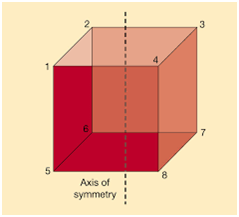
Where,
a and b are coefficients of x2 and x respectively.
c is a constant term.
Plane symmetry
A space figure will have plane symmetry, when it is divided into two halves by a plane and each half is the reflection of the other across the plane. The plane is called a plane of symmetry of the space figure.
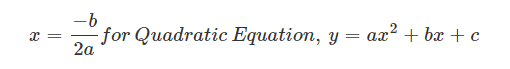
Fig: Space figure showing plane symmetry
It should be noted that not all the planes of reflection can be the planes of symmetry, although the plane shown below cuts the rectangular prism into two equal halves, it is not a plane of symmetry. When a vertex A for the prism is reflected across the plane the image A’ is viewed, that is not mapped on the other half of the prism, this becomes true for all the points that lies in the prism excluding the ones that intersect with the plane.
Fig: Plane of symmetry in a rectangular prism
Multiple planes of symmetry
Multiple planes of symmetry can be observed in, for example, the rectangular prism below has three planes of symmetry.
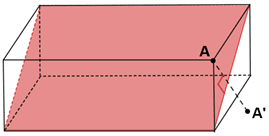
Fig: Three planes of symmetry in a rectangle
A cube shows nine planes of symmetry, six planes of symmetry are diagonal while three planes of symmetry are parallel to the surfaces.
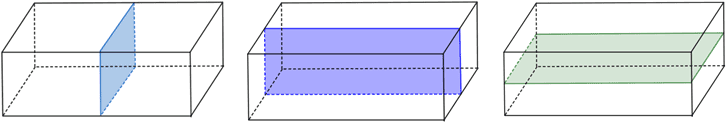
Fig: Nine planes of symmetry in a cube
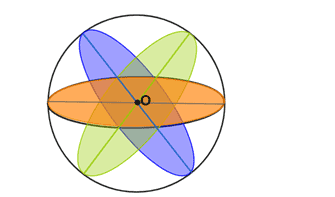
A sphere contains infinitely many planes of symmetry. If a plane contains the centre point, O, of the sphere, it is a plane of symmetry for the sphere. Three of the infinitely many planes of symmetry are shown in the sphere below.
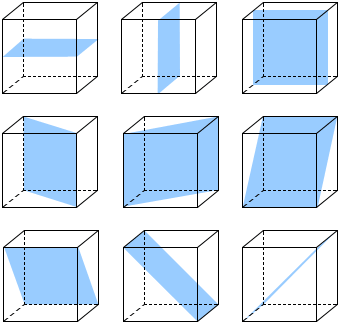
Fig: A sphere showing planes of symmetry
Types of magnetic behaviours
All substances are known to show magnetic behaviour of some kind, as they are made up of charged particles namely electrons and protons, the magnetic properties are determined by the way the groups of these atoms behave themselves and also the way in which electron cloud arrange themselves, the atom (or group of atoms) in effect becomes a magnetic dipole or a mini bar magnet that can align according to the magnetic field applied. The net effect of all these dipoles determines the magnetic properties of the Magnetic Materials. In reality, every atom is magnetic. However, there are different types of atomic magnetism–and these magnetic effects get even more complicated when atoms are arranged in crystal structures. Magnetism is a quantum-mechanical effect that is related to electron spin
Magnetic materials are defined by their response to an external field (in other words, their permeability).
There are 3 main types of magnetic materials: ferromagnetic, paramagnetic, and diamagnetic.
Para magnetism
Para magnetism is a magnetism, here many objects get attracted by an externally applied magnetics field.is a kind of magnetism where several objects are attracted through an externally applied magnetic field.
The materials that exhibit paramagnetic are mostly chemical elements and some compounds as they possess magnetic permeability greater than or equal to 1. The magnetic moment induced by the applied field is linear to the strength and weakness of the field. It usually needs a sensitive analytical balance to detect the effect and different modern measurements on paramagnetic materials that are often conducted with a SQUID magnetometer.
The paramagnetic material becomes good magnetic material when placed in the strong magnetic material. They act as a magnet that attracts and repels other magnetic materials. When the magnetic field is removed, the total magnetic alignment of the magnetic dipoles is lost and the dipole returned to their normal random motion. This condition is known as Para magnetism.
The Super Para magnetism is described as the property it is noticed that the magnetic moments are changed in some magnetic materials, they change their direction and behaves like a paramagnet event. According to Curie law, if there is no magnetic field applied, curie temperature is applied at the same time as they show high magnetic susceptibility.
Curie’s Law
According to this law, the magnetization in the paramagnetic material is inversely proportional to the temperature, which means the more the temperature of the paramagnetic material increases, its magnetization decreases.
M = C(B/T)
Where,
C = Curie constant,
T = temperature in Kelvin and,
B = applied the magnetic field.
Diamagnetism
Diamagnetic materials usually repel from a magnet. Technically, these solids create an induced magnetic field in a direction opposite to an externally applied magnetics force and are repelled by the applied magnetic field. This phenomenon is just the opposite behaviour exhibited by paramagnetic materials.
Diamagnetism is a very weak form of magnetism that is induced by a change in the orbital motion of electrons due to an applied magnetic field. This magnetism is non permanent and persists only in the presence of an external field. The magnitude of the induced magnetic moment is very small, and its direction is opposite to that of the applied field.
The orbital motion of electrons present on the atoms of diamagnetic solids produces magnetic fields as it creates tiny atomic current loops. When an external magnetic force is applied to a material, these current loops tend to align in such a way as to oppose the applied field.
In diamagnetic materials, there is no permanent net magnetic moment per atom as all the electrons are paired. Due to the influence of an external magnetic force, diamagnetic properties arise from the realignment of the electron paths. Most elements in the periodic table like copper, silver, and gold, are diamagnetic in nature. Sebald Justinus Brugmans discovered diamagnets in the year 1778. Michael Faraday further demonstrated that diamagnetism or magnetism is a property of a matter and every material or solid reacts accordingly.
Ferromagnetism
It is a physical phenomenon that occur in certain material like iron have strong attraction toward each other, the ferromagnets occur in very rare earth materials and gadolinium, one of the common phenomena that is met in life that is responsible for magnetism in magnets.
One of the vital requirements of ferromagnetic material is that ions and atoms should possess permanent magnetic moments. Some ions and atoms consist of the permanent magnetic moment that may be considered as a dipole that comprises a north pole separated from a south pole.
Ferromagnetism or the meaning of ferromagnetism is a mechanism through which certain materials form permanent magnets. With the aid of a strong electrostatic field, these materials can be permanently magnets. With the aid of a strong electrostatic field, these materials can be permanently magnetized. Ferromagnetic metal ions are grouped into small regions called solid-state domains. So, every domain is acting like a tiny magnet. The domains of a ferromagnetic unmagnetized piece are randomly oriented so that their magnetic moments are cancelled out. When this material is put in a magnetic field, all domains are oriented in the direction of the magnetic field, creating a powerful magnetic effect. Also, when the magnetic field is withdrawn and the ferromagnetic material becomes a permanent magnet, this order of domains remains the same. There are many different forms of magnetism, but ferromagnetism is of the strongest form and is responsible for the widespread occurrence of magnetism in magnets experienced in everyday life.
Some degree of dipole alignment can be witnessed has there existed a large atomic magnetic moment. This type of magnetic arrangement can be found in some elements such as iron, cobalt, nickel, and their alloys.
Properties of Ferromagnetic materials
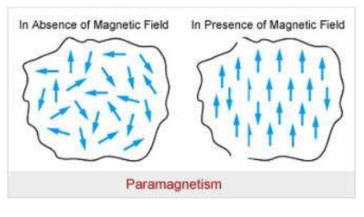
Dependence of Para magnetism on S & L
Two contributions to the electronic magnetic moment
An orbital magnetic moment due to orbital angular momentum
A spin magnetic moment due to electro spin

Total angular moment J and the total magnetic moment are not collinear, however, in an external field, mJ+s processes fast about J and J processes much slower than Hz, thus the time average component of the magnetic moment (m) = mII is parallel to J.
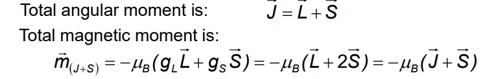
Coupling of spin and orbital moments yields the total angular momentum of electrons
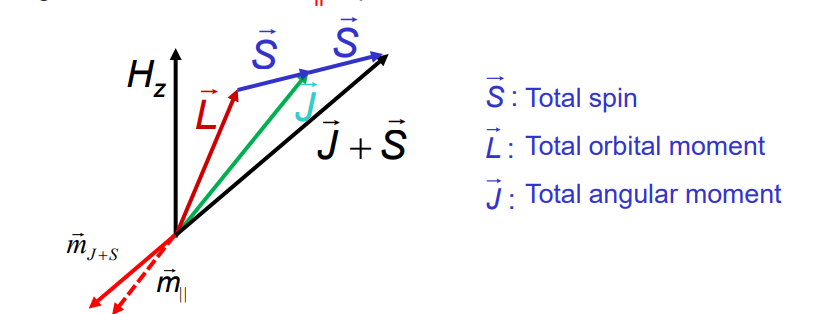
The spin-orbit (so) interaction or LS-coupling is described by:
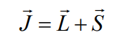
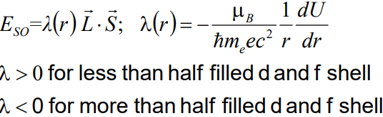
Coupling of Spin orbital moments is due to Zeeman-splitting of spin magnetic moment in the magnetic field that is produced by orbital moment.
Volumetric analysis is a quantitative analytical method that is used widely, as indicated by its name, the method involves the measurement of a solution of known concentration that is utilised to estimate the concentration of the analyte, this process is called titration and the solution in the burette is called the titrant.
The analyte of unknown concentration reacts with a chemical of unknown amount in the presence of an indicator (mostly phenolphthalein) to show the end-point. The amount of unknown chemical in the specific volume of solution is determined by the mole fraction of the equation.
The accuracy in weighing these substances is the key to accurate results, indicators are necessary to establish the endpoint in a volumetric analysis. Acid-Base titrations, Redox titrations and Complexometric titrations are the major techniques in volumetric analysis
It is commonly used to determine the unknown concentration of a known reactant. Volumetric analysis is often referred to as titration, it’s a laboratory method where one substance is of known concentration and volume is used to react with another substance of unknown concentration
Basic principles of volumetric analysis
The solution to be analysed contains an unknown amount of chemicals. The reagent of unknown concentration reacts with a chemical of an unknown amount in the presence of an indicator (mostly phenolphthalein) to show the end-point
It involves the estimation of a substance in solution by neutralization, precipitation, oxidation or reduction by means of another solution of accurately known strength. This solution is known as standard solution.
Volumetric analysis depends on measurements of the volumes of solutions of the interacting substances. A measured volume of the solution of a substance A is allowed to react completely with the solution of definite strength of another substance B. The volume of B is noted. Thus, we know the volume of the solutions A and B used in the reaction and the strength of solution B; so, the strength of the other solution A is obtained. The amount (or concentration) of the dissolved substance in volumetric analysis is usually expressed in terms of normality. The weight in grams of the substance per litre of the solution is related to normality of the solution as,
Weight of the substance (g per litre) = Normality × gram equivalent weight of the substance.
Precipitation Titration (Ag++)
When a slightly soluble precipitate is formed from a titration reaction the titration is known as precipitation titration. The most important precipitation process in titrimetric analysis utilizes silver nitrate as the reagent (Argentometric process).
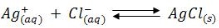
There are several methods to determine the endpoints of these reactions, but the most important method is the formation of coloured precipitate is taken as priority.
The chloride concentration is initially high and AgCl is the less soluble salt, therefore AgCl gets precipitated, as soon as the chloride ions are completed the addition of small quantity of silver nitrate solution brick red colour silver chromate becomes visible. The titration should be carried out in neutral solution or in very faintly alkaline solution. i.e., within the pH range 6.5-9. In acid solutions following reaction occurs.
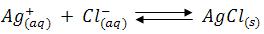
Consequently, the chromate ions concentration is reduced and the solubility product of silver chromate may not be exceeded. In markedly alkaline solution, silver hydroxide (Ksp = 2.3 x 108) might be precipitated.
2. The titration can be carried out with dichlorofluorescein as the indicator. Dichlorofluorescein is an adsorption indicator, having the property to change colour when they stick to the surface of a precipitate. During the titration the dichlorofluorescein molecules exist as negatively charged ions (anions) in solution. During the method as the Agcl precipitate is formed, the excess Cl- ions in the solution form a layer of negative charge on the precipitate surface. As the equivalence point is reached and passed, the excess Cl- ions on the precipitate surface are replaced by excess Ag+ ions, giving the surface a positive charge. The negatively charged indicator will be attracted to the positively charged precipitate surface where it absorbs and changes colour. The suspended precipitate will have a pink tinge because of some premature displacement of chloride ion by the dichlorofluorescein ion. When the pink colour starts to persist for slightly longer periods of time, the drip rate is lowered. The end point is reached when the entire solution turns pink. It is important that the AgCl precipitate be prevented from coagulation during the titration. For this reason, a small amount of dextrin is added to the solution.
Iodometric titration (Cu++)
Iodine is a mild oxidizing agent. In the presence of a suitable reducing agent, it is reduced to iodine ion, I-. In addition to this, all oxidizing agents having electrode potential greater than 0.54 V can oxidize I- to I2. When iodine solution is directly used for the estimation of reducing agents, the titration is called iodometric titration (iodimetry). The titrations involving the iodine liberated in a chemical reaction are called iodometric titration (iodometry).
The given solution of copper sulphate reacts with KI in the presence of acetic acid, resulting in the formation of dark brown, Copper (II) iodide compound
 CuSO4+KI CuI2 + K2SO4 + 5H2O
CuSO4+KI CuI2 + K2SO4 + 5H2O
(Copper (II)
iodide)
Copper (II) iodide is an unstable compound and will further form
 2CuI2 Cu2 I2 + I2
2CuI2 Cu2 I2 + I2
(Copper (I)
iodide)
The iodine that is liberated is titrated with sodium thiosulphate solution (Standard solution) till a yellow colour appears, in this process freshly prepared starch is the indicator, this freshly prepared starch is added to the solution until a blue colour appears
 Starch + I2 Starch-I2 (Blue coloured complex compound)
Starch + I2 Starch-I2 (Blue coloured complex compound)
The solution is further titrated with Sodium thiosulphate solution till a milky white precipitate appears
 2Na2S2O3 + I2 Na2 S4O3 + 2NaI
2Na2S2O3 + I2 Na2 S4O3 + 2NaI
(Sodium tetrathionate
Milky white)
Acid Base titration (Ca++)
Water hardness can be readily determined by titration method with the chelating agent, EDTA (ethylenediaminetetraacetic acid) (Greek χηλή, chelè, meaning claw). This reagent is a weak acid that can lose four H (in bold) on complete neutralization.
The four acid oxygen sites and the two nitrogen atoms have unshared pair of electrons, which can form bonds with a metal ion forming a complex ion or coordination compound. The complex that is formed is quite stable, and the conditions of its formation can easily be controlled so that it is ready for selection for a particular metal ion. when a titration to determine the concentration of a metal ion is carried out, the added EDTA quantitatively combines with the cation to form the complex. The endpoint is reached when essentially all of the cation has reacted. In this analysis a solution of EDTA will be standardize by titration against a standard solution made from calcium carbonate, CaCO3. The EDTA solution can then be used to determine the hardness of an unknown water sample. Since both EDTA and Ca2+ are colourless, therefore a special indicator is used to detect the end point of the titration. The indicator mostly used is called Eriochrome Black T, which forms a very stable wine-red complex, MgIn–, with the magnesium ion. A small amount of this complex will be present in the solution during the titration. As EDTA is added, the complex free Ca2+ and Mg2+ ions, leaving the MgIn– complex alone until essentially all of the calcium and magnesium are converted to chelates. At this point EDTA concentration will increase marginally to displace Mg2+ from the complex indicator; the indicator reverts to its uncombined form, which is sky blue, thus establishing the end point of the titration. The titration is carried out at a pH of 10, in a NH3/NH4 + buffer, the equations for the reactions which occur during the titration are:


 Titration reaction: HY3–(aq) + Ca2+(aq) CaY2–(aq) + H+ (aq) (also for Mg2+) End point reaction: HY3–(aq) + MgIn– (aq) MgY2–(aq) + HIn2–(aq)
Titration reaction: HY3–(aq) + Ca2+(aq) CaY2–(aq) + H+ (aq) (also for Mg2+) End point reaction: HY3–(aq) + MgIn– (aq) MgY2–(aq) + HIn2–(aq)
As the indicator requires a trace of Mg2+ to operate properly, a little magnesium ion will be added to each solution. The effect of the added Mg2+ can be subtracted by titrating a blank.
A simple way to calculate the hardness in water.
A sample of hard water contains 1mg cacl2 and 1 mg mgcl2 per
litre. calculate the hardness of water in terms of caco3 present in per 106 parts of water.
Hardness of water in terms of Caco3= (molar mass of hardness causing substance*molecular
weight of Caco3)/ (molecular weight of hardness causing substance.
Thus, cacl2 hardness = 1mg*100/111 = 0.900ppm for Mgcl2
Hardness = 1mg*100/95=1.052ppm
Total hardness of water in terms of Caco3 = 0.90+1.052= 1.952ppm
Gravimetric analysis utilises the measurement of mass concept where the amount of an analyte (the ion being analysed) can be determined. The method depends on the masses of the two compounds that contain the analyte and comparing their weight. The principle behind gravimetric analysis is that the mass of an ion in a pure compound can be determined and then used to find the mass percent of the same ion in a known quantity of an impure compound. In order for the analysis to be accurate, certain conditions must be met:
Gravimetric analysis is the most dependant and most accurate analytical methods that are available.it involves weighing of substance, the element or radical to be estimated is converted into a stable compound with definite composition and the mass of the compound is determined accurately, from this, the mass of element or radical is calculated.
The gravimetric analysis involves a) precipitation b) filtration c) washing of the precipitate and d) drying, ignition and weighing of the precipitate.
Following are the four fundamental types of gravimetric analysis:
The Gravimetric Estimation of Nickel:
The nickel is precipitated as nickel dimethyl glyoxime by adding alcoholic solution of dimethyl glyoxime C4H6(NOH)2 and then adding a slight excess of aqueous ammonia solution.
When the pH is buffered in the range of 5 to 9, the formation of the red chelate occurs quantitatively in a solution. The chelation reaction occurs due to donation of the electron pairs on the four nitrogen atoms, not by electrons on the oxygen atoms. The reaction is performed in a solution buffered by either an ammonia or citrate buffer to prevent the pH of the solution from falling below 5. If the pH does become too low the equilibrium of the above reaction favours the formation of the nickel (II) ion, causing the dissolution of Ni (DMG)2 back into the mother liquor.
A slight excess of the reagent has no action on the precipitate, but a large excess should be avoided because of the possible precipitation of the reagent itself. The precipitate formed is soluble in mineral free acid, it is therefore important to prevent the addition of excess reagent as it may crystallise out as chelate.it should be noted that the complex is slightly soluble in alcoholic solutions and the addition of chelating agents will reduce these errors to some extent. The amount of the reagent added is also governed by the presence of other metals such as cobalt, which form soluble complexes with the reagent. If a high quantity of these ions is present, a greater amount of DMG must be added. The nickel dimethylglyoximate is a very bulky precipitate. Therefore, the sample weight used in the analysis must be carefully controlled to allow more convenient handling of the precipitate during the transfer to the filtering crucible. The compactness of the precipitate is improved by adjusting the pH to 3 or 4, followed by the addition of ammonia solution.
The pH increases gradually in the concentration of ammonia resulting in a complex precipitate, which is dense. After the completion of collecting and drying the precipitate, the nickel content of the solution is calculated stoichiometrically from the weight of the precipitate.
The structure of DMG & the complex with nickel ions is given below;
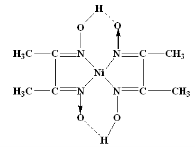
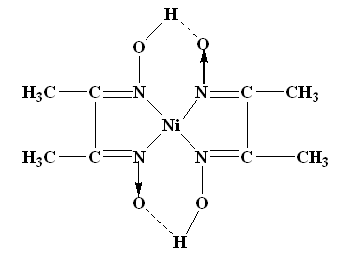
The Gravimetric Estimation of Barium:
The given barium chloride solution is made up to a definite volume. A measured volume of it is then treated with dilute sulphuric acid and then treated with dilute sulphuric acid and barium precipitated as barium sulphate.
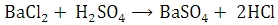
The precipitated barium sulphate is separated and weighed. The mass of Barium in the whole of the given solution is calculated knowing that 233.36 g of barium sulphate contains 137.36 g of barium.
The Gravimetric Estimation of Sulphate (SO4++)
 Principle: the method provides the most precise results and is highly recommended for sulphate concentrations above 10 mg/mL. The sulphate ions in the sample form a precipitate by the addition of barium chloride solution to water sample acidified with hydrochloric acid and kept near the boiling point.
Principle: the method provides the most precise results and is highly recommended for sulphate concentrations above 10 mg/mL. The sulphate ions in the sample form a precipitate by the addition of barium chloride solution to water sample acidified with hydrochloric acid and kept near the boiling point.
SO42- + Ba2+ BaSO4.
In highly alkaline water maintained near the boiling temperature, BaCO3 may get precipitated and eliminate this, the sample needs to be acidified. Excess barium chloride is used to completely precipitate the sulphate ions. The precipitate of BaSO4 is highly insoluble and therefore there is a possibility for the precipitate to form colloidal condition that is difficult to remove by filtration procedures. To facilitate the conversion of colloidal form to crystalline form, the samples at temperatures near the boiling point for a few hours are digested.
Procedure: A water sample of about 200ml is transferred into a beaker, to these two drops of methyl red indicator is added followed by dropwise addition of Conc. HCl till the colour of the solution changes to pink. Two drops of conc. HCl are added in excess. The volume of the solution is reduced to 50ml by heating it to boiling, with stirring until a white precipitate is formed. Two drops of hot BaCl2 are added in excess. The precipitate (BaSO4) is digested for about 2 hours (or until the precipitate becomes settles down). It is filtered using Whatman filter paper No. 42 (ash less filter paper), quantitatively. The precipitate is washed several times with distilled water until the washings are free of chloride ions. After it completely drains out, the filter paper along with the precipitate is transferred carefully into previously weighed Gooch crucible and ignited at 800-9000C till all traces of filter paper are burnt. The crucible is cooled in a desiccator and finally weighed. Let the amount of BaSO4 precipitated be W g.
Calculations: We know that, 233.4 g of BaSO4 contains 96.06 g of sulphate.
Therefore, W g of BaSO4 contains (96.06 x W)/233.4 g of sulphate.
That is, 200 mL of water sample contains (96.06 x W)/233.4 g of sulphate.
Therefore 1000 mL of water sample contains (96.06 x W x 1000)/ (233.4 x 200) g of sulphate
The Gravimetric Estimation of Copper (Cu++)
We will analyse an alloy containing copper to determine its percent content in it. Copper will be precipitated as CuSCN (solubility product Kso=12.7). This means that Cu2+ ions will be reduced to Cu+ before they are precipitated using SCN-.
 2Cu2+ + HSO3 - + H2O 2Cu+ + HSO4 - + 2H+
2Cu2+ + HSO3 - + H2O 2Cu+ + HSO4 - + 2H+
 Cu+ + SCN- CuSCN
Cu+ + SCN- CuSCN
This is an excellent method, since most thiocyanates of other metals are soluble, in particular thiocyanates of Bi, Cd, As, Sb, Sn, Fe, Ni, Co, Mn and Zn. In presence of moderate amounts of Bi, Sb or Sn it is desirable to add 2-3 g of tartaric acid to prevent of hydrolysis of their salts. Solubility of CuSCN increases with pH, so excessive amounts of ammonium ions should be absent., as should also oxidizing agents. The solution should be only slightly acidic, since the solubility of CuSCN increases with decreasing pH because of complexing ability of thiocyanate anions. In this analysis, Pb, Hg, Se, Te and precious metals ions interfere and contaminate the precipitate. Consequently, the conditions of experiment are as follows: Slight acidity with respect to HCl than H2SO4. The presence of a reducing agent, for instance H2SO3 or NH4HSO3, to reduce Cu (II) to Cu(I). A slight excess of NH4SCN. A large excess increases the solubility of the copper thiocyanate due to formation of a complex. The absence of oxidizing agents. The precipitate is curdy and readily coagulates by boiling. It is washed with dilute ammonium thiocyanate with an addition of H2SO3 or NH4HSO3, to avoid oxidization of Cu(I).
The final report should contain masses of empty and full filters, calculated masses of the precipitated CuSCN, calculations made to achieve the final result, and – if necessary – the brief analysis of the results. The result is mass of elemental copper in each sample (in grams) and the averaged weight percent of Cu in the alloy or mineral under examination. The ratio of molecular masses of Cu to CuSCN is 0.5225.
The Gravimetric Estimation of Magnesium (Mg++)
The reaction between Mg2+ ions and EDTA can be represented like this.
Mg2+ + H2Y2- → MgY2- + 2H+
Prepare a standard solution of magnesium sulfate and titrate it against the given EDTA solution using Eriochrome Black T as the indicator. A buffer solution is prepared for maintaining the pH of about 10. When the reaction is complete all the magnesium ions would have been complexed with EDTA and the free indicator would impart a blue color to the solution. Estimation of magnesium ions using edta.
Estimation of magnesium ions in the given sample: 20 mL of the given sample of solution containing magnesium ions is pipetted into a 250 Erlenmeyer flask, the solution is diluted to 100 mL, warmed to 40 degrees C, 2 mL of a buffer solution of pH 10 is added followed by 4 drops of Eriochrome black T solution. The solution is titrated against the standardized EDTA solution.
The precipitate formed is filtered and washed continually till traces are removed the sample is dried and weighed.
The proton number for different isotopes of an element remains unchanged.
All isotopes are not radioactive, stable isotopes never decay or decay gradually. Radioactive isotopes undergo decay.
When an isotope decays, the starting material is the parent isotope. The resulting material is the daughter isotope
Detection and Separation.
For all types of radiations, can be exposed to Photographic film where it is used to monitor the exposure of working personnel with high energy emitters A visible track in a cloud or bubble chamber can pick up radioactivity, as can a calorimeter if the energy emitted is quite high.
The problems faced with the methods of detection is the energy of the emitter must be high enough to travel some distance through air. Many nucleotides that emit particle, don’t travel significantly through air even for a short distance especially 14-C and 3-H labelled compounds, posing danger to the proximity of molecules like DNA. For example, a beta particle emitted by tritium cannot enter a sheet of paper, yet it can be hazardous if present in the body fluids, A liquid scintillation detector can pick up radiation from "soft" beta emitters as well as from other radioisotopes. A particle that ionises is passes through a crystal or liquid phosphor, that absorbs energy and produces flashes of light on re-emission. Usually, the emitter must be dissolved in liquid containing the luminescent compound, so that the distance travelled by the particle is very short.
The kinetic energy in beta particles is different from one decay to the next. Every isotope has a specific energy spectrum with a range of energies that can be predicted. Typical energy spectra for different in shape and magnitude, as with the commonly used 14-C and 3-H.
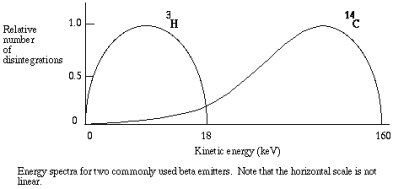
In liquid scintillation counting, the material that contains the radioisotope is dissolved in an organic solvent containing an aromatic solute (the scintillant). When radioactive decay takes place, the energy of a beta particle is transferred by collision to an electron in the shell of the scintillant, exciting that electron. The electron then returns to its ground state, releasing a photon. However, the transfer of energy can either be from beta particle to solvent to scintillant or to the scillant directly, usually there can be multiple collisions from each beta particle The number of photons emitted following each atomic disintegration is proportional to the energy of the released beta particle.
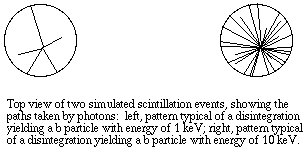
The vial is lowered into a dark chamber with photoelectric detectors on each side. Each "flash" received by the detectors corresponds to one atomic disintegration. The detectors are connected through photomultiplier tubes and then to a microprocessor unit that records not only each event, but also the number of photons detected during each event (brightness of the flash). At brief intervals, such as 1/100 second, the instrument calculates the number of flashes per unit time, displaying them as counts per minute, or CPM.
Methods of Separation
Diffusion
The diffusion method is dependent on the point that at thermal equilibrium ,two isotopes with similar energy will have different average velocities, the lighter atoms or the molecules can travel more quickly and can easily diffuse through a membrane, the difference in speed is proportional to the square root of the mass ratio, resulting in a small separation and many stages have to be introduced to achieve high purity, the method is expensive as work is needed to push the gas through the membrane and many stages are required for the process to take place.
The first large scale separation of Uranium isotopes was obtained by the United States, in the large plant set up for separation, they used uranium hexafluoride gas as the process fluid. Nickel powder and electro-deposited nickel mesh diffusion barriers were pioneered by Edward Adler and Edward Norris
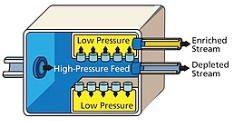
Fig: Gaseous diffusion uses semi-permeable membrane to separate enriched uranium
Centrifugal
It involves the rapid rotation of the material that allows the heavier isotope to move closer to the outer radial wall, this is often done with the Zippe-type centrifuge.
Fig: A Cascade of gas centrifuges at the uranium enrichment plant, US
In 1919 Aston and Lindemann suggested the centrifugal separation of isotopes and the first successful experiment was reported by Beams and Haynes on isotopes of chlorine in 1936. However, attempts to use the technology during the Manhattan project were unproductive.in present day it is one of the widely used methods to enrich uranium and the process is kept a secret to hinder its widespread. In general, a feed of UF6 gas is connected to a cylinder that is rotated at high speed. Near the outer edge of the cylinder heavier gas molecules containing U-238 collect, while molecules containing U-235 concentrate at the centre and are then fed to another cascade stage, the use of gaseous technology centrifugally to enrich isotopes is favoured as they reduce power consumption to a great extent when compared to the other conventional techniques such as diffusion plants since fewer cascade steps are required to reach similar degrees of separation. In fact, gas centrifuge using uranium hexafluoride have largely replaced gaseous diffusion technology for uranium enrichment. As well as requiring less energy to achieve the same separation, far smaller scale plants are possible, making them an economic possibility for a small nation attempting to produce a nuclear weapon. Pakistan is believed to have used this method in developing its nuclear weapons.
Vortex tubes are used in South Africa for the Vortex separation process, the gas is injected tangentially into a chamber with special geometry that further increases its rotation to a very high rate, causing the isotopes to separate. The method is simple because vortex tubes have no moving parts, but energy intensive, about 50 times greater than gas centrifuges. A similar process, known as jet nozzle, was created in Germany, with a demonstration plant built in Brazil, and they went as far as developing a site to fuel the country's nuclear plants.
Electromagnetic

Fig: Schematic diagram of uranium isotope separation in a calutron
This method is like mass spectroscopy, and is also called by its name. it uses the concept that charged particles get deflected in the presence of a magnetic field and the amount of deflection depends on the particle as the output is extremely low though the expenditure for the quantity produced is expensive, but high purity levels are achieved. This method is often used for processing small amounts of pure isotopes for research or specific use (such as isotopic tracers), but is impractical for industrial use.
electromagnetic separation for much of the uranium used in the first United States atomic bomb (Manhattan project). the device used in this principle is called Calutrons. The method was restricted as impractical after the war. It had only been undertaken (along with diffusion and other technologies) to guarantee there would be enough material for use, whatever the cost. Its main eventual contribution to the war effort was to further concentrate material from the gaseous diffusion plants to even higher levels of purity.
Laser
In this method a laser is set to a wavelength which excites one isotope of the material and ionises those atoms specially. The resonant absorption of light for an isotope is dependent on its mass and certain hyperfine interactions. between electrons and the nucleus, allowing finely tuned lasers to interact with only one isotope. After the atom is ionized it can be removed from the sample by applying an electric field. This method is often abbreviated as AVLIS (atomic vapour laser isotope separation). This method has only recently been developed as laser technology has improved, and is currently not used extensively. However, it is a major concern to those in the field of nuclear proliferation because it may be cheaper and more easily hidden than other methods of isotope separation. Tuneable lasers used in AVLIS include the dye laser and more recently diode lasers
Tracer technique and applications
A radioactive isotope acts in a similar way as a normal isotope and can be present in the organism. But the concentration and the location of the radioactive isotope can be easily determined by measuring the radiation emitted. A radioactive isotope acts as an indicator or tracer that makes it possible to follow the course of a chemical or biological process. The method to be followed is to substitute the radioactive atoms with the stable atoms of same kind in a substance and then to follow the "tagged" atoms with the help of radiation detector in the process.
Traces are applicable in the agricultural field to study the taking up of a fertilizer by a plant. For example, if a plant is given radioactive carbon-14 then the plant releases "beta radiation" and thus by measuring radioactivity in different parts of the plant. The method also helps in expanding the complex processes involved in photosynthesis.
The tracer technique is useful to detect faults in underground pipes.
Traces are widely used in medicine to analyse the process of digestion and the movement of chemicals in our body. Few organs in our body absorb some chemicals. For example, "radio_iodine"is absorbed mostly by the thyroid gland, phosphorus by bones and cobalt by liver, these can serve as a tracer, however the doses of radiation knees to be carefully controlled otherwise the radiation could do more damage than help.
Tracer technique is used to substitute radioactive atoms for the ones that are stable atoms and are of same kind in a substance. these then to follow the "tagged" atoms with the help of radiation detector in the process.
A radioactive isotope is known to behave in a similar manner as a normal isotope does when inside a living organism. The location and concentration of a radioactive isotope is known to be easily determines as it can measure the emitted radiations. These isotopes are used in chemistry and biochemistry to help understand chemical reactions and biological interactions.
Radiocarbon Dating
Radiocarbon dating was invented in the 1950s by the American chemist Willard F. Libby and a few of his students at the University of Chicago: in 1960, he won a Nobel Prize in Chemistry for the invention. It was the first absolute scientific method ever invented: that is to say, the technique was the first to allow a researcher to determine how long ago an organic object died, it is still the best and most accurate of dating techniques devised.
How Does Radiocarbon Work?
All living things exchange the gas Carbon 14 (C14) with the atmosphere around them, animals and plants exchange Carbon 14 with the atmosphere, fish and corals exchange carbon with dissolved C14 in the water. Throughout the life of an animal or plant, the amount of C14 is perfectly balanced with that of its surroundings. When an organism dies, that equilibrium is broken. The C14 in a dead organism slowly decays at a known rate: its "half-life".
The half-life of an isotope like C14 is the time it takes for half of it to decay away: in C14, every 5,730 years, half of it is gone. So, if you measure the amount of C14 in a dead organism, you can figure out how long ago it stopped exchanging carbon with its atmosphere. Given relatively pristine circumstances, a radiocarbon lab can measure the amount of radiocarbon accurately in a dead organism for as long as 50,000 years ago; after that, there's not enough C14 left to measure.
Tree Rings and Radiocarbon
There is a problem, however. Carbon in the atmosphere fluctuates with the strength of earth's magnetic field and solar activity. You have to know what the atmospheric carbon level (the radiocarbon 'reservoir') was like at the time of an organism's death, in order to be able to calculate how much time has passed since the organism died. What you need is a ruler, a reliable map to the reservoir: in other words, an organic set of objects that you can securely pin a date on, measure its C14 content and thus establish the baseline reservoir in a given year.
Fortunately, we do have an organic object that tracks carbon in the atmosphere on a yearly basis: tree rings. Trees maintain carbon 14 equilibrium in their growth rings and trees produce a ring for every year they are alive. Although we don't have any 50,000-year-old trees, we do have overlapping tree ring sets back to 12,594 years. So, in other words, we have a pretty solid way to calibrate raw radiocarbon dates for the most recent 12,594 years of our planet's past.
But before that, only fragmentary data is available, making it very difficult to definitively date anything older than 13,000 years. Reliable estimates are possible, but with large +/- factors.
Key takeaway
The symmetry of a molecule provides a great deal of information about its quantum states, even without a detailed solution of the Schrödinger equation. A geometrical transformation which turns a molecule into an indistinguishable copy of itself is called a symmetry operation. Magnets form when ferromagnetic or ferrimagnetic materials are exposed to an electromagnetic field. Magnets display certain characteristics: There is a magnetic field surrounding a magnet. Magnets attract ferromagnetic and ferrimagnetic materials and can turn them into magnets. A magnet has two poles that repel like poles and attract opposite
References




